Generating a periodic system for FHI-aims (sub system)
Objective: Turn a sub system of the MD system into a geometry file suitable for FHI-aims to be simulated with periodic boundary conditions.
Specifically, we want to select a box around the solute that extends 5 Å in all directions and only include the solvent molecules in this buffer zone together with the solute.
Danger
Unlike when we were exporting the full MD system, which was simulated under periodic boundary conditions, here we are cutting out a orthorhombic sub-system that is not truly periodic. We are assuming that the solvent is sufficiently homogeneous so that the newly introduced periodic boundaries do not introduce substantial artifacts such as clashes due to wrapped atoms or incorrect local coordination geometry. In practice, such a reduced system will likely have clashes and require additional work to be suitable for running in FHI-aims.
Despite these potential problems with intermolecular interactions, the protocol described here will ensure that intramolecular bonds are correctly wrapped around the new unit cell. The resulting system should be carefully examined and if necessary, additional steps such as energy minimization with the classical force field under the new periodic boundary conditions are necessary. Below we sketch out an example protocol for how to use GROMACS to do such an energy minimization to remove clashes.
Warning
This tutorial assumes that the MD trajectory was simulated inside a orthorhombic unit cell (i.e., all angles 90°); the simple approach shown here will not work correctly for non-orthorhombic unit cells. (Although MDAnalysis works with all unit cells, the simple cutting and rewrapping step used here requires orthorhombic unit cells.)
Protocol
The following commands can be used interactively in the python or
ipython interpreter or a Jupyter notebook. Alternatively, combine
them in a script extract_reduced_pbc.py and run the
script with python extract_reduced_pbc.py.
Set-up, Centering, Trajectory frame selection
Import NumPy, MDAnalysis and the MDAnalysis transformations module:
Load the topology (TPR) and trajectory (TRR) of the classical MD
simulation into the universe u:
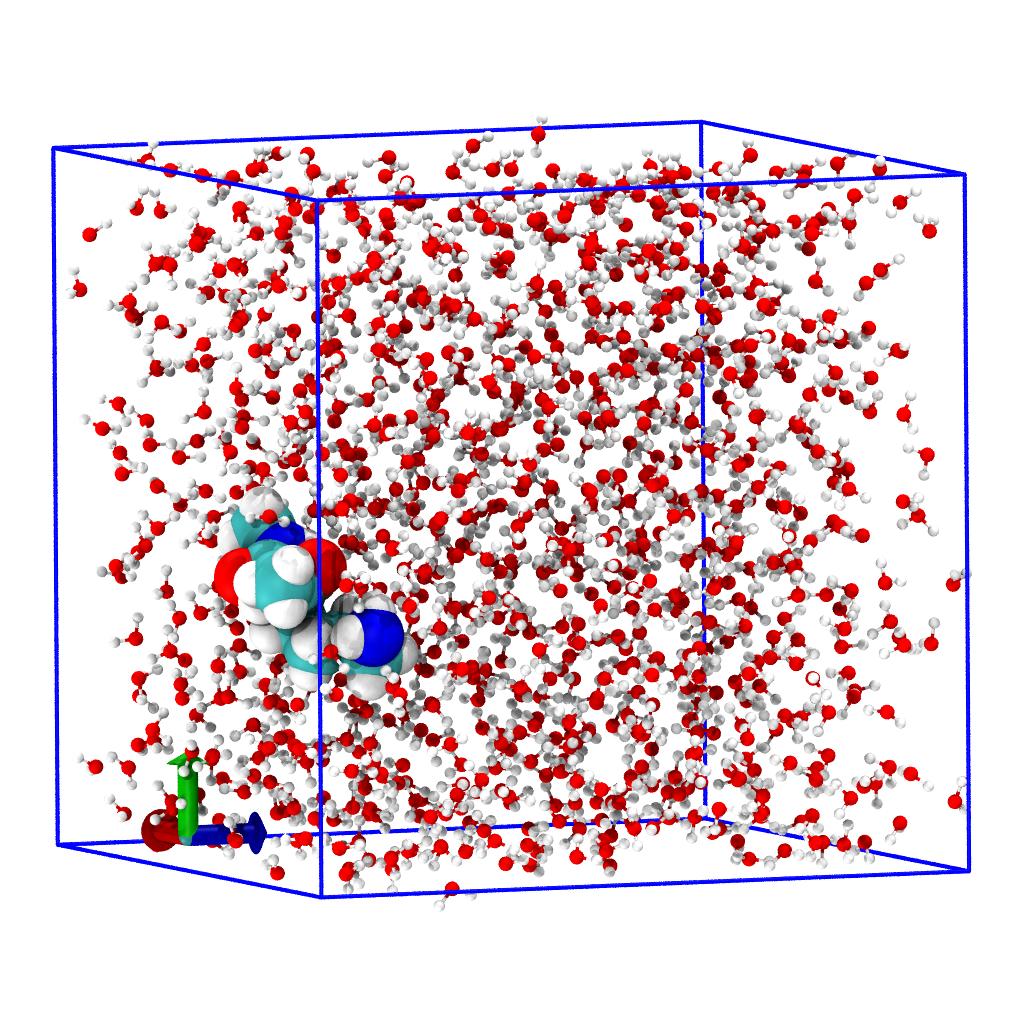
The following steps are the same as in the Set-up step for the full MD system (and following steps) so we will only summarize them here.
- Change atom names to elements.
- Create AtomGroup instances for the water molecules (
water) and the solute by using selection by residue name and checking that we selected everything. - Center the system.
- Move to the last frame of trajectory.
u.atoms.names = u.atoms.elements
water = u.atoms.select_atoms("resname SOL")
solute = u.atoms.select_atoms("resname ACE LYSN NME")
assert u.atoms == solute + water
workflow = [transformations.unwrap(u.atoms),
transformations.center_in_box(solute, center='geometry'),
transformations.wrap(water, compound='atoms')]
u.trajectory.add_transformations(*workflow)
u.trajectory[-1]
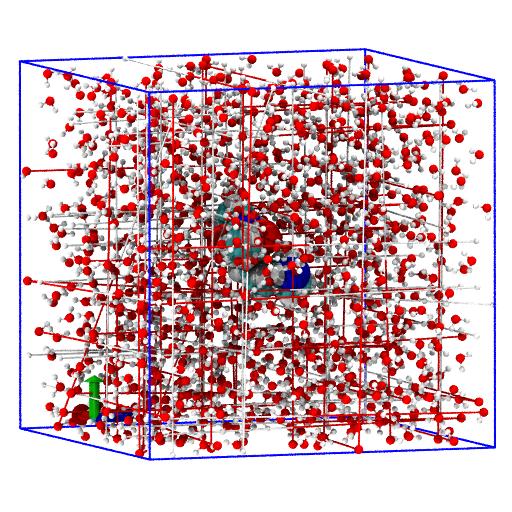
Find new reduced unit cell
Solute bounding box
We first find the bounding box of the solute (the orthorhombic cell
with axes parallel to the coordinate system that exactly fits the
solute in its current orientation) with the
AtomGroup.bbox()
method:
The variable bbox = [[xmin, ymin, zmin], [xmax, ymax, zmax]] contains the
lower and upper corner of the enclosing bounding box.
For our information we compute the lengths A, B, C of the
bounding box and compare them to the current dimensions of the unit
cell.
A, B, C = bbox[1] - bbox[0]
print("unit cell: a={uc[0]:7.4f} Å b={uc[1]:7.4f} Å c={uc[2]:7.4f} Å ⍺={uc[3]}° β={uc[4]}° ɣ={uc[5]}° ".format(uc=u.dimensions))
print(f"solute bbox: A={A:7.4f} Å B={B:7.4f} Å C={C:7.4f} Å")
Warning
If the angles of the unit cell are not all equal to 90° then stop here. This protocol will only work for orthorhombic unit cells.
For the example files the output is
unit cell: a=31.0358 Å b=31.0358 Å c=31.0358 Å ⍺=90.0° β=90.0° ɣ=90.0°
solute bbox: A= 7.8396 Å B= 7.3349 Å C= 7.0937 Å
New orthorhombic unit cell
We add a distance of
in all directions (extend the lower corner of the bounding box by-delta and the upper
corner by +delta). rmin now contains the coordinates of the lower
left front corner of the new unit cell (centered on the solute) and rmax
are the coordinates of the upper right back corner.
Tip
Change delta to fit your own requirements. Visually check the
reduced system once you write it out after the last step. Make
sure that there are no clashes and that there is a sufficient
solvation shell around the solute; "sufficient" is determined by
the scientific question that you want to answer.
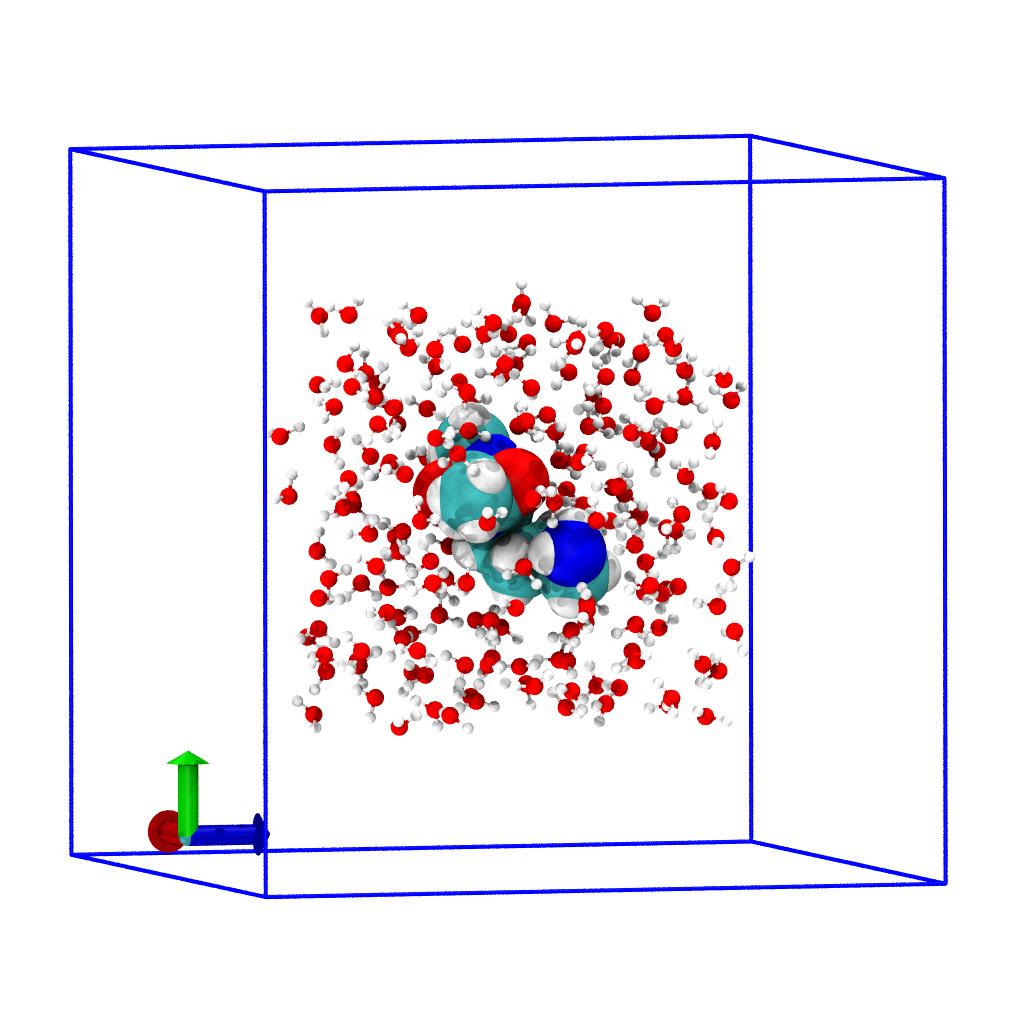
Select water in new unit cell
We select complete molecules with the byres selection
keyword inside the extended box around the solute and
use a boolean and selection with the prop selection keyword to
find all water atoms that have their coordinates
within the limits of our delta-expanded bounding box:
water_box = water.select_atoms(f"byres prop x > {rmin[0]} and prop x < {rmax[0]} and "
f"prop y > {rmin[1]} and prop y < {rmax[1]} and "
f"prop z > {rmin[2]} and prop z < {rmax[2]}")
Create combined system with new unit cell
Combine solute and the reduced water system into our reduced sub-system:
The dimensions of the new orthorhombic unit cell are [A, B, C, 90,
90, 90]:
In MDAnalysis, unit cells are always oriented in such a way that the unit cell vectors originate in \((0, 0, 0)\), that the first box vector is parallel to the Cartesian \(x\) axis \((1, 0, 0)^T\), the plane spanned by the first and second unit cell vector lies in the Cartesian \(xy\) plane, and the third vector should be oriented into the positive-\(z\) half-space to form a right-handed coordinate system.
In the next steps we will apply coordinate transformations to make our new reduced system conform to the unit cell requirements of MDAnalysis.
Note
All coordinate transformations that follow are temporary in that
they change the current system coordinates but they will all be
overwritten and forgotten when the trajectory is moved to another
frame (e.g. u.trajectory[2]); we will write out the currently
altered coordinates to save them (Alternatively, we could use
in-memory trajectories to make changes permanent but
that is a more advanced topic.)
The new unit cell will have lower left corner (0,0,0) so lets shift
all coordinates of the system accordingly; this will ensure that our
small system (which is still part of the full system u.atoms) will
be positioned so that its lower left front corner will be located at
the origin:
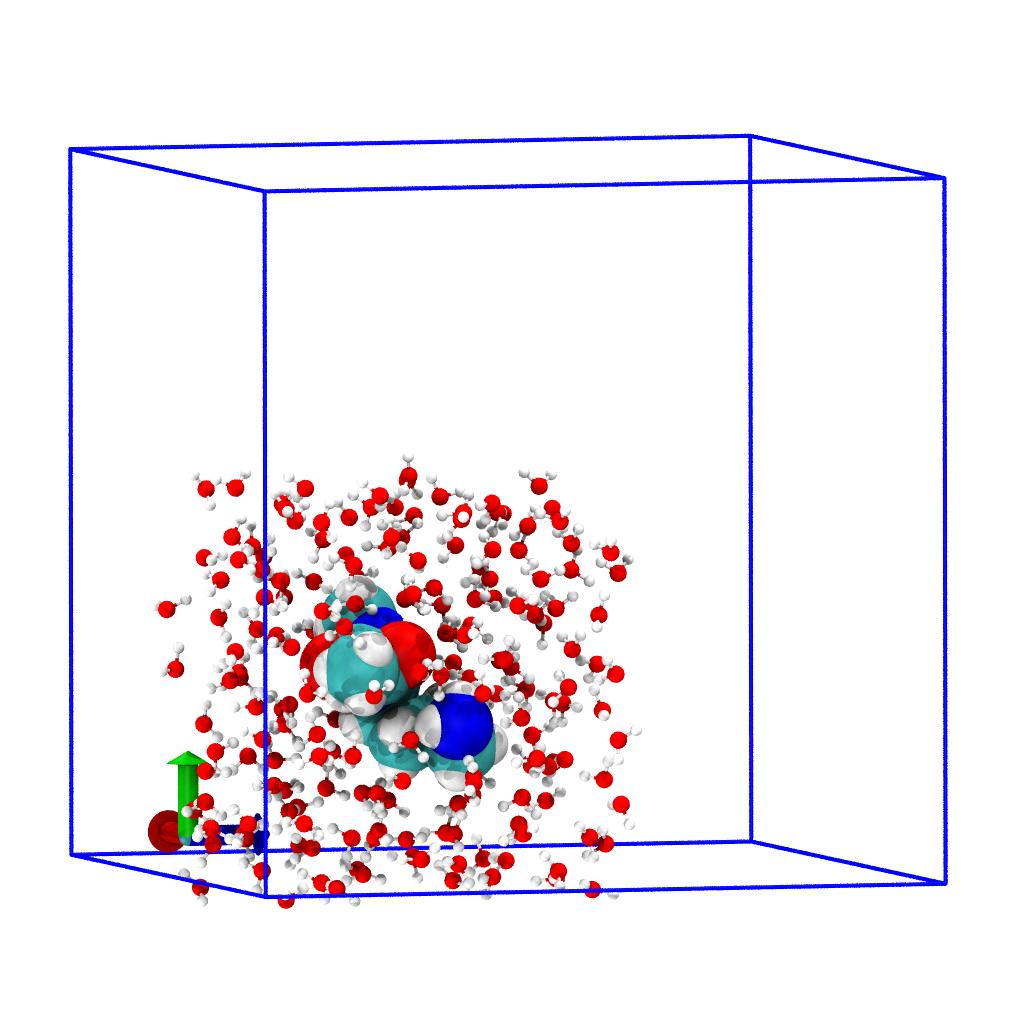
Set new unit cell dimensions and wrap atoms
Set the new unit cell dimensions
so that the correct unit cell is written to output.Note on how .dimensions is really changed globally.
This command actually sets dimensions for the whole Universe because
.dimensions is really a property of the trajectory and is thus shared
between all AtomGroups of the same Universe. Like the changing of
positions, this change is only temporary and active only as long as
the trajectory frame is not changed.
Then we also need to put all atoms inside the new unit cell newbox using
AtomGroup.wrap()
inplace=True we change the coordinates of all atoms in
smaller_system directly.
We now have cut out an orthorhombic subsystem and adjusted the unit cell information so that FHI-aims will be able to work with the system.
Output to geometry.in
We want to write the our reduced system to a FHI-aims geometry.in
file (MDAnalysis supports reading and writing the FHI-aims file
format). We take the atoms of our sub-system from the
smaller_system.atoms attributes and use its write() method (which
can determine the output format from the filename extension):
We can also write a file in PDB format for visualization in tools that may not understand the FHI-aims format:
(Ignore harmless warnings about attributes that are set to default values to satisfy the PDB standard.)Suppressing warnings in Python
You can temporarily silence the warnings with the warnings filter:
Complete script
Save this script as extract_reduced_pbc.py:
# -*- coding: utf-8 -*-
import warnings
import numpy as np
import MDAnalysis as mda
from MDAnalysis import transformations
u = mda.Universe("./files/MD/NPT/md.tpr", "./files/MD/NPT/md.trr")
u.atoms.names = u.atoms.elements
water = u.atoms.select_atoms("resname SOL")
solute = u.atoms.select_atoms("resname ACE LYSN NME")
assert u.atoms == solute + water
workflow = [transformations.unwrap(u.atoms),
transformations.center_in_box(solute, center='geometry'),
transformations.wrap(water, compound='atoms')]
u.trajectory.add_transformations(*workflow)
u.trajectory[-1]
# bounding box of the peptide
bbox = solute.bbox()
A, B, C = bbox[1] - bbox[0]
print("unit cell: a={uc[0]:7.4f} Å b={uc[1]:7.4f} Å c={uc[2]:7.4f} Å ⍺={uc[3]}° β={uc[4]}° ɣ={uc[5]}° ".format(uc=u.dimensions))
print(f"solute bbox: A={A:7.4f} Å B={B:7.4f} Å C={C:7.4f} Å")
# additional distance in all directions
delta = 5
rmin = bbox[0] - delta
rmax = bbox[1] + delta
# select complete molecules with "byres" inside the extended box around
# the solute
water_box = water.select_atoms(f"byres prop x > {rmin[0]} and prop x < {rmax[0]} and "
f"prop y > {rmin[1]} and prop y < {rmax[1]} and "
f"prop z > {rmin[2]} and prop z < {rmax[2]}")
# combined system
smaller_system = solute + water_box
# new unit cell: A, B, C, alpha, beta, gamma
newbox = np.concatenate([rmax - rmin, [90, 90, 90]])
# Put all atoms inside the new unit cell
u.atoms.translate(-rmin)
smaller_system.wrap(compound="atoms", box=newbox, inplace=True)
smaller_system.dimensions = newbox
smaller_system.write("geometry.in")
with warnings.catch_warnings():
# write PDB output without the needless warnings about defaults for the PDB file
warnings.simplefilter("ignore", UserWarning)
smaller_system.write("geometry.pdb")
Adjust the input files and the selection criteria and then run it with
Energy minimization for the reduced system
The resulting geometry.in is probably not yet suitable as input for
FHI-aims due to steric clashes.
Finding clashes
We can find all interatomic distances in the small_system below a
cutoff (say, 2 Å) with MDAnalysis using the fast
MDAnalysis.lib.distances.self_capped_distance() function:
import numpy as np
from MDAnalysis.lib import distances
pairs, dist = distances.self_capped_distance(
smaller_system, max_cutoff=2,
box=smaller_system.dimensions, return_distances=True)
dist contains the distance for each pair of atoms in the
pair list pairs, taking the periodic boundaries into account via the
box=smaller_system.dimensions argument.
A histogram of the distances shows us immediately that there are a number of contacts \(< 0.8\) Å (which is much closer than a typical bond length involving hydrogen).
Plotting the distance histogram
The distance histogram can be immediately obtained from dist
with numpy/matplotlib:

In this example, the number of atom pairs that are clashing are
The explicit distances are
>>> dist[dist < 0.8]
>>> array([0.68607039, 0.76228015, 0.45474766, 0.73319411, 0.71172214,
0.69234899, 0.41190428, 0.7202629 , 0.58936537, 0.49754758,
0.51289614, 0.53577986, 0.66571993])
We can find the pairs with
>>> pairs[dist < 0.8]
>>> array([[340, 229],
[424, 181],
[423, 181],
[182, 425],
[203, 62],
[587, 556],
[585, 556],
[585, 555],
[590, 69],
[590, 71],
[491, 411],
[491, 412],
[411, 489]])
where each tuple consists of the atom indices of the two atoms. We can
also directly index the AtomGroup smaller_system with these indices
to get more information; here we just print to quickly convince
ourselves that these are distances between different water molecules
(indicated by different residue IDs ("resids")) and therefore these
are almost certainly clashes that we introduced:
>>> clashes = [smaller_system[pair] for pair in pairs[dist < 0.8]]
>>> print(clashes[0])
>>> <AtomGroup [<Atom 1475: H of type HT of resname SOL, resid 484 and segid seg_1_SOL>, <Atom 815: H of type HT of resname SOL, resid 264 and segid seg_1_SOL>]>
>>> [clashgroup.resids for clashgroup in clashes]
>>> [array([484, 264]),
array([678, 195]),
array([678, 195]),
array([195, 678]),
array([224, 31]),
array([948, 885]),
array([948, 885]),
array([948, 885]),
array([955, 36]),
array([955, 36]),
array([757, 661]),
array([757, 661]),
array([661, 757])]
Removing clashes with energy minimization
One can take different approaches to reduce/remove the clashes. Here we use the classical force field approach again and do an energy minimization ("geometry optimization") with the GROMACS MD program.
Note
In order to do this part of the tutorial you need to have a working GROMACS installation as described in Running the classical MD simulations.
The main considerations are
-
The unit cell is so small that the normal cut off distances for the neighbor list and non-bonded cutoffs are too large. We have to reduce them to values smaller than half the box size. Under normal circumstances we would not use such small values, especially as the force fields are parametrized with specific values for the VDW/LJ interactions. For the purpose of removing clashes, these are acceptable compromises.
-
To better capture the physics of the small periodic system we use Ewald VDW (PME VDW) in GROMACS, which computes LJ in the periodic system without a cutoff. This is generally more expensive and the CHARMM force field was parameterize with LJ cutoffs so normally we would use cutoffs at the literature values. If PME LJ is not available then LJ with short cutoffs should also work as we are primarily concerned with close contacts.
-
We will use two separate energy minimizations:
-
First with no constraints on any bonds to allow all water molecules to easily rearrange. Water molecules were simulated with the rigid TIP3P water model which has all interatomic distances fixed with SETTLE constraints. By replacing constraints with bonds for this step, we give the molecules more freedom to relax clashes without running into numerical instabilities when constraints cannot be easily satisfied. We use a moderate convergence criterion (
emtol) of maximum force of 1000 kJ/mol/nm = 1.036 eV/Å. We can use the excellent L-BFGS minimizer for this step. -
In the second step we enable constraints again to pull water molecules into shape. We use a simple and robust steepest descent minimizer but now use a fairly strict (for classical force fields) convergence criterion of 100 kJ/mol/nm = 0.104 eV/Å.
-
Directory set up
In the following we work in a directory EnergyMinimization. Create
two separate work directories
We use the geometry.pdb file that we wrote out previously. Copy it
to the topology directory together with all topology files that were
necessary for Running the classical MD
simulations
topol.top: topology file (overall system information)aceKnme.itp: included topology file (information for the lysine dipeptide)posre.itp: position restraints (not needed but referenced intopol.top)
Info
All input files are also downloadable from files/EnergyMinimization.
Update topology file
Edit the topol.top file to update the number of water ("SOL")
molecules. Get the number with water.n_residues (in Python) or
grep '^ATOM.*SOL.*O' geometry.pdb | wc -l (in the shell). Your
molecules section should look like
First energy minimization without constraints
We run an initial energy minimization with the L-BFGS minimizer and
distable all constraints by using constraints = None and selecting
water topology files for "flexible" water, i.e., where bonds are not
constrained with SETTLE (-DFLEXIBLE). The important lines in the MDP
file 01_emin_noconstraints.mdp are
As mentioned above, all cutoffs need to be shortened to less than half the box lengths so that the non-bonded section looks like
MDP parameters
For further details on setting simulation parameters see Molecular dynamics parameters (.mdp options) in the GROMACS manual.
The MDP file for this minimization step can be downloaded from 01_emin_noconstraints.mdp.
We now generate the portable binary run input file for GROMACS with
the gmx grompp command
gmx grompp -maxwarn 1 -f 01_emin_noconstraints.mdp -c ../topology/geometry.pdb -p ../topology/topol.top -o 01_emin.tpr
where we use our geometry.pdb as input for the starting coordinates
and 01_emin_noconstraints.mdp sets the run parameters. We have to
use -maxwarn 1 to ignore one harmless warning about mismatching atom
names (because we renamed atoms to elements for FHI-aims).
We can then run the minimization itself with gmx mdrun
The L-BFGS minimizer only runs in serial so we set the number of
threads to 1 (-nt 1) for gmx mdrun. The run should produce output
files 01_emin.{gro,tpr,log,edr} within seconds. The log file should
indicate that the minimization converged:
Low-Memory BFGS Minimizer converged to Fmax < 1000 in 43 steps
Potential Energy = -6.5631802e+03
Maximum force = 8.5441931e+02 on atom 5
Norm of force = 2.6728458e+02
Second energy minimization with constraints
In the second energy minimization step we enable constraints for bonds involving hydrogens again (which are solved with SETTLE for water and LINCS for everything else) and use a tighter convergence criterion. The run input MDP file 02_emin_constraints.mdp contains
Generating the TPR file and running the minimization follows the same protocol as before,
gmx grompp -maxwarn 1 -f 02_emin_constraints.mdp -c 01_emin.gro -p ../topology/topol.top -o 02_emin.tpr
gmx mdrun -nt 4 -deffnm 02_emin -s 02_emin.tpr
Note that the starting coordinates are read from the output of the
previous energy minimization, 01_emin.gro. The minimization can be
run in parallel and 4 threads (-nt 4) should make this task finish
in a couple of seconds. The output should show convergence, e.g.,
Steepest Descents converged to Fmax < 100 in 1524 steps
Potential Energy = -9.8530654e+03
Maximum force = 9.2342125e+01 on atom 5
Norm of force = 1.4365285e+01
The final energy minimized coordinates are in the 02_emin.gro file.
Check energy minimized geometry for clashes
We analyze the interatomic distances for clashes (\(d < 0.8\) Å) as
before, but we now load the final energy minimized coordinates in the
02_emin.gro file. Load the final coordinates into a MDAnalysis
Universe and calculate the distances:
em = mda.Universe("EnergyMinimization/energy_minimization/02_emin.gro")
pairs, dist = distances.self_capped_distance(em.atoms, max_cutoff=2,
box=em.dimensions, return_distances=True)
How many distances are below our clash distance?
Zero! This indicates success, we removed all clashes.Convert GROMACS output to AIMS geometry.in
We use MDAnalysis to convert the coordinates in 02_emin.gro to a
geometry.in file as before:
import MDAnalysis as mda
TPR = "02_emin.tpr"
GRO = "02_emin.gro"
AIMSIN = "geometry.in"
PDB = "geometry.pdb"
u = mda.Universe(TPR, GRO)
u.atoms.names = u.atoms.elements
u.atoms.write(AIMSIN)
u.atoms.write(PDB, remarks="energy minimized with GROMACS")
The resulting geometry.in file should now be suitable for
calculations in AIMS. In particular, the density SCF iterations should
converge.