Generating a periodic system for FHI-aims (full system)
Objective: Turn the full periodic MD system into a geometry file suitable for FHI-aims to be simulated with periodic boundary conditions.
Tip
The current tutorial exports the complete classical MD system and introduces the basic steps that we will also use when we are exporting a subsystem.
Protocol
The following commands can be used interactively in the python or
ipython interpreter or a Jupyter notebook. Alternatively, combine
them in a script extract_all_pbc.py and run the
script with python extract_all_pbc.py.
Set-up
Import MDAnalysis and the transformations module:
Load the topology (TPR) and trajectory (TRR) of the classical MD
simulation into the universe u:
Change atom names to elements because these names are written to the output file and FHI-aims wants elements, not force field atom names:
Note
With TPR files, information is available for elements. With other input files, MDAnalysis will have to guess the elements.
Create AtomGroup instances for the water molecules (water) and the
capped lysine (solute) by using selection by residue name
We can check that we partitioned the whole system into solute and
water with a simple assertion that the combined AtomGroup of solute +
water is the same as all the atoms in the system:
If the assertion fails then you should check your selections.
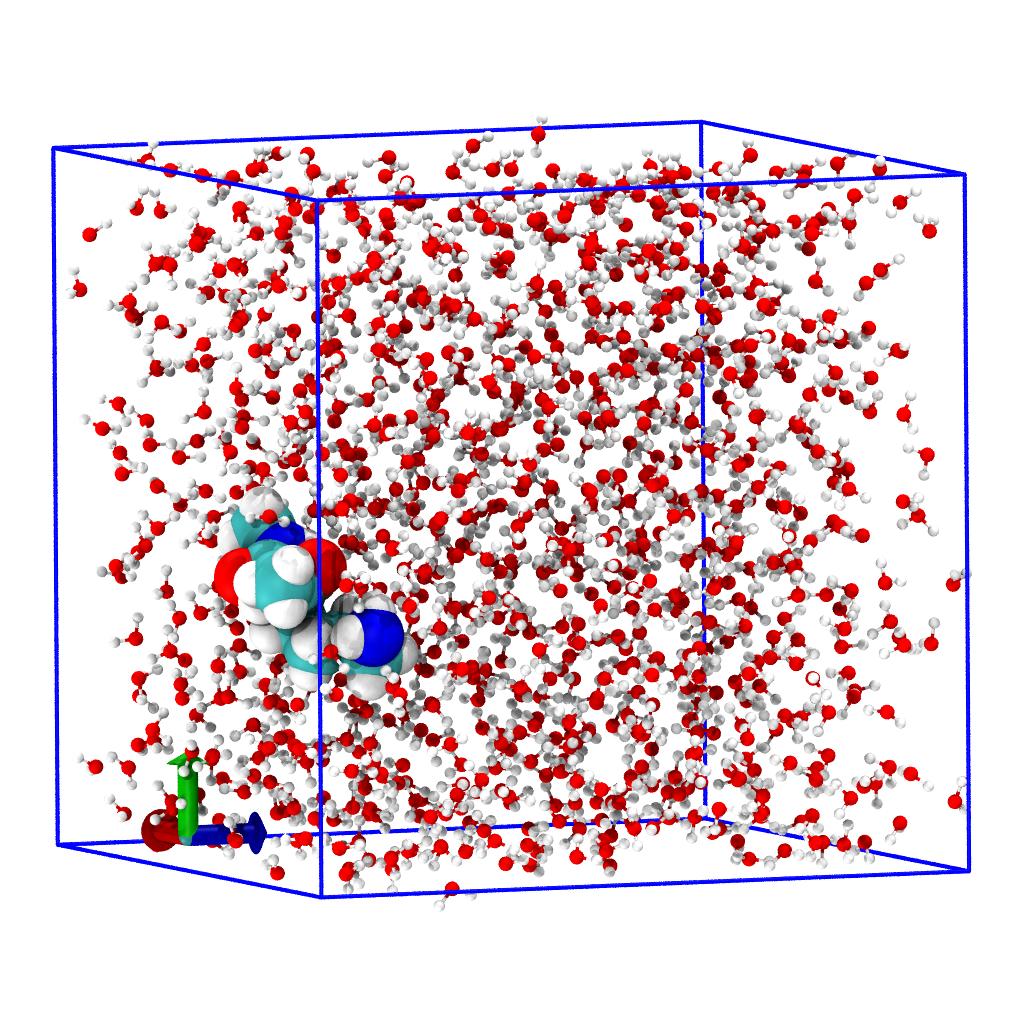
Centering
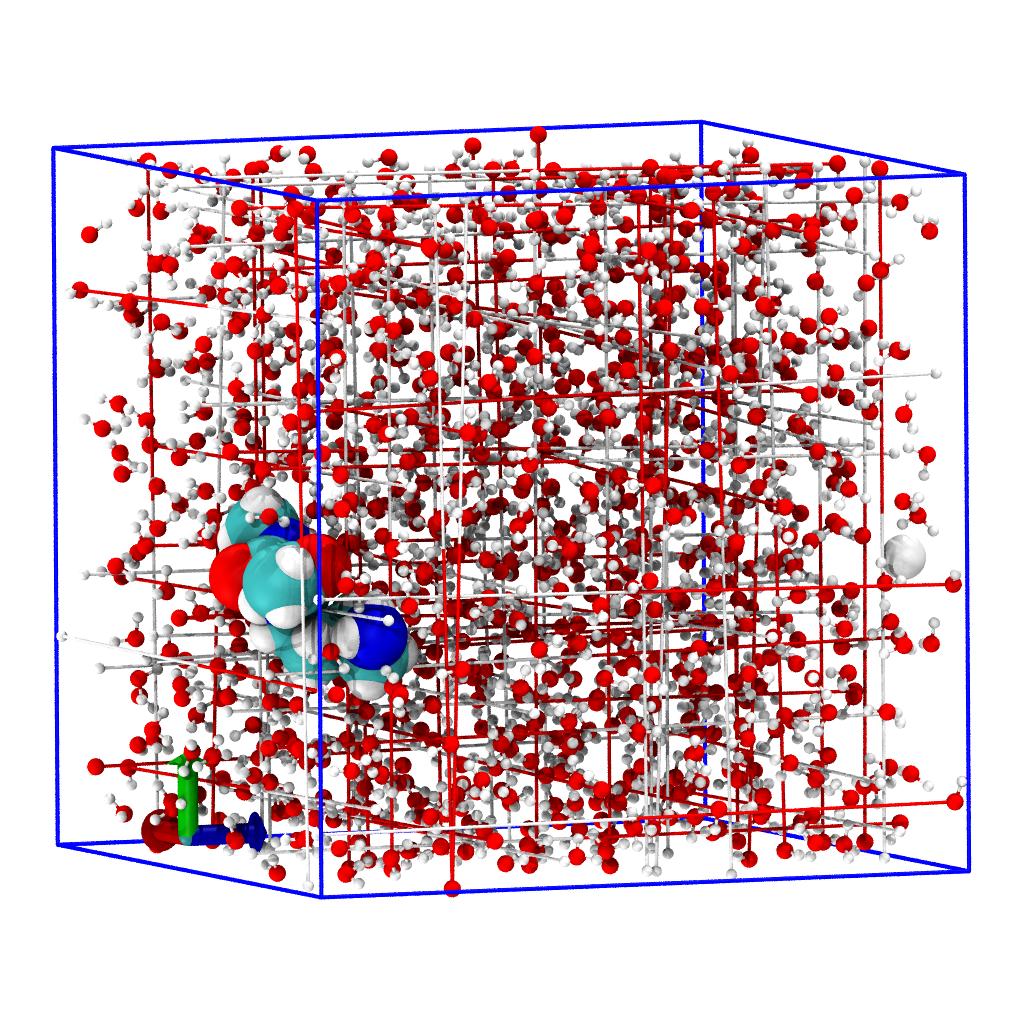
The objective is to generate a trajectory where
- the peptide is at the center of the simulation cell
- water is packed around the peptide
- water molecules are within periodic boundaries.
We use on-the-fly transformations to rewrite the coordinates of the trajectory while reading the TRR file from the disk. We create a workflow of functions that is run between reading the coordinates from file and storing them in the Timestep instance for the rest of MDAnalysis to see.
workflow = [transformations.unwrap(u.atoms),
transformations.center_in_box(solute, center='geometry'),
transformations.wrap(water, compound='atoms')]
u.trajectory.add_transformations(*workflow)
unwrap(u.atoms)unwraps all molecules over the box boundaries if necessary.center_in_box(solute, center='geometry')shifts all coordinates so that the center of the box coincides with the center of geometry of the AtomGroupsolute(which we selected previously).wrap(water, compound='atoms')ensures that allwateratoms are inside the box that was centered on the solute in transformation 2.
We then add the workflow to the u.trajectory instance and activate
it for all future use of the universe instance.
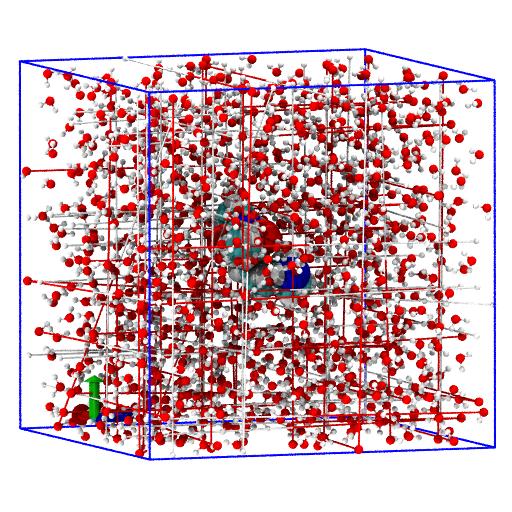
Select trajectory frame
MDAnalysis keeps track of the current frame of the trajectory. At the beginning, the frame "cursor" points to frame 0.
Info
MDAnalysis uses zero-based indexing for frames.
You can move to any frame between 0 and u.trajectory.n_frames - 1 by indexing the u.trajectory
instance, i.e., u.trajectory[frame].
To move to the last frame of trajectory use
All AtomGroups will now read data from the last trajectory frame (suitably transformed).
Output to geometry.in
We want to write the our system to a FHI-aims geometry.in
file (MDAnalysis supports reading and writing the FHI-aims file
format). We simply take all atoms of the system from the u.atoms
attribute of the Universe:
We can also write a file in PDB format for visualization in tools that may not understand the FHI-aims format (ignore warnings about attributes that are set to default values to satisfy the PDB standard):
Suppressing warnings in Python
You can temporarily silence the warnings with the warnings filter:
Complete script
Save this script as extract_all_pbc.py:
import MDAnalysis as mda
from MDAnalysis import transformations
u = mda.Universe("./files/MD/NPT/md.tpr", "./files/MD/NPT/md.trr")
u.atoms.names = u.atoms.elements
water = u.atoms.select_atoms("resname SOL")
solute = u.atoms.select_atoms("resname ACE LYSN NME")
assert u.atoms == solute + water
workflow = [transformations.unwrap(u.atoms),
transformations.center_in_box(solute, center='geometry'),
transformations.wrap(water, compound='atoms')]
u.trajectory.add_transformations(*workflow)
u.trajectory[-1]
u.atoms.write("geometry.in")
u.atoms.write("geometry.pdb")
Adjust the input files and the selection criteria and then run it with